

Mass Spectrometry
Over the last decade, mass spectrometry has evolved as the method of choice for identification and quantification of proteins and other biomolecules. The high sensitivity, mass resolution and speed of modern instruments allows for comprehensive analysis of complex mixtures (i.e. several thousand protein species) in even small amounts of biological samples (microgram range) when coupled to chromatographic pre-separation. Analytes are ionized and then separated by their mass-to-charge (m/z) ratio either in intact and/or fragmented form. Different setups have been developed with distinct specifications and applications.
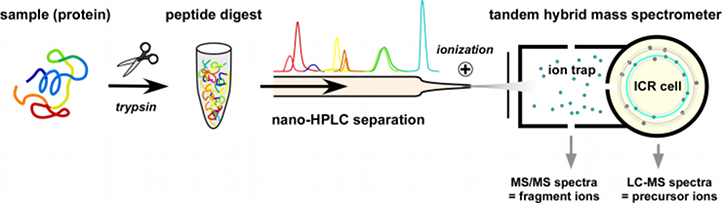
This combination allows for efficient parallel acquisition of fragment ion spectra (MS/MS) for identification and full precursor ion spectra (MS) which provide quantitative information.
Our laboratory uses three hybrid tandem mass spectrometers containing a linear ion trap for collection of ions and ion fragmentation (highest sensitivity and speed) coupled to a detector (orbitrap or ion cyclotron resonance (ICR) cell) with high mass resolution and mass precision. This combination allows for efficient parallel acquisition of fragment ion spectra (MS/MS) for identification and full precursor ion spectra (MS) which provide quantitative information. Before entering the spectrometer, samples are separated by nanoflow liquid chromatography (nano-LC; Ultimate 3000) on a reversed-phase capillary column.
Protein identification
- Single MS/MS spectrum and total sequence coverage of BK alpha splice variants and phosphorylated sites from a series of rat brain APs

Intact proteins are usually too large for efficient LC-MS analysis with standard instrumentation and are therefore digested into peptides beforehand. Trypsin is most widely used because it is efficient, sequence specific and generates a high number of suitable peptides (i.e. with an m/z between 370 and 1200). Still, the resulting sample complexity is high: each protein gives rise to several tens to hundreds of ion species. In practice, only a fraction of these ions is detected, fragmented and matched with information in the database. Crucial parameters for protein identification by MS sequencing are the overall sample complexity, the resolution and capacity of the nano-LC, the sampling speed, sensitivity and mass accuracy of the spectrometer. In our optimized setups, several hundreds of proteins are routinely identified per sample run (at less than µg loads, sensitivity of ≤1 fmol), with halfmaximal peptide elution peak widths of 30s, data-dependent acquisition of up to 10 MS/MS spectra per second, and precursor mass accuracies of < 3 ppm. This allows for high-confidence identification of even small and low abundant proteins in complex samples and high sequence coverage with detailed information on splice variants and post-translational modifications.
Protein quantification


Quantification of proteins is essential for determination of purification specificity, as well as protein-protein interaction stability, stoichiometry and selectivity. Different techniques have been established, most of them involving chemical or metabolic labelling with stable isotopes but also having limitations with regard to dynamic range, multiplexing and use of native tissues. Thus, we employ label-free quantification, which is based on extraction of ion signal intensities and their integration over time and m/z (peak volume, PV). Relative quantification using PV ratios of suitable (i.e. selected by correlation analysis) peptides provides high sensitivity and a broad linear range (at least 3-4 orders of magnitude) as actually required for quantitative evaluation of affinity purifications versus controls. To determine molar amounts of different proteins (absolute quantification), we use standards consisting of fused protein domains or concatenated specific peptides as equimolar calibrants. The high accuracy of this approach allows for elucidation of complex stoichiometry.
 Previous:
Membrane Proteomics
Previous:
Membrane Proteomics
